
Bharath Ramsundar, Ph.D.
Education
-
B.S. Electrical Engineering and Computer Science and Mathematics, 2011
Departmental Citation (Valedictorian) for Mathematics, Class of 2012
Highest Honors in EECS and Mathematics
University of California Berkeley -
Ph.D. Computer Science, Hertz Fellow, 2018
Thesis Supervisor: Dr. Vijay Pande
Thesis Title: “Molecular Machine Learning with DeepChem”
Stanford University
About Bharath
Bharath received a BA and BS from UC Berkeley in EECS and Mathematics and was valedictorian of his graduating class in mathematics. He did his PhD in computer science at Stanford University where he studied the application of deep-learning to problems in drug-discovery. At Stanford, Bharath created the deepchem.io open-source project to grow the deep drug discovery open source community, co-created the moleculenet.ai benchmark suite to facilitate development of molecular algorithms, and more. Bharath's graduate education was supported by a Hertz Fellowship, the most selective graduate fellowship in the sciences. After his PhD, Bharath co-founded Computable a startup that built better tools for collaborative dataset management. Bharath is currently the founder and CEO of Deep Forest Sciences, which is building an AI-powered suite for drug and materials design and discovery.
Bharath is the lead author of "TensorFlow for Deep Learning: From Linear Regression to Reinforcement Learning", a developer's introduction to modern machine learning, with O'Reilly Media, and "Deep Learning for the Life Sciences". Additionally, he authored "The DeepChem Book" in collaboration with the DeepChem team, published in 2024, and is currently working on "Differentiable Physics: Machine Learning for Physical Systems".
Awards and Honors
- Hertz Fellowship, 2012-2018
- Departmental Citation (Valedictorian) for UC Berkeley Mathematics, 2012
- Highest Honors in EECS and Mathematics, 2012
- UCRegents’ and Chancellor’s Scholar, 2007
Books
- "Differentiable Physics: Machine Learning for Physical Systems". In preparation.
- "The DeepChem Book"
- "TensorFlow for Deep Learning: From Linear Regression to Reinforcement Learning"
- "Deep learning for the life sciences"
Talks
2024

Conference
Building Scientific Computing Infrastructure Software with the PyTorch Ecosystem

Lecture
Chemical Foundation Models for Drug Discovery

Lecture
Intro to ML in Drug Discovery: Principles & Applications
2022

Seminar
DeepChem: Towards Open Source Foundations for Modern Drug Discovery

Podcast
Open-Source Drug Discovery with DeepChem with Bharath Ramsundar
2021

Podcast
Applications of AI

Webinar
ChemBERTa | Scientific Machine Learning Webinar

Podcast
Deep Learning in the Sciences
2020

Podcast
Deep Learning for drug discovery, founding an AI startup & more

Podcast
Deep Learning for Molecules and Medicine Discovery
2018

Presentation
Decentralized Data Markets In Theory And Practice
2016

Presentation
Democratizing Drug Discovery with DeepChem
Papers and Patents
2026
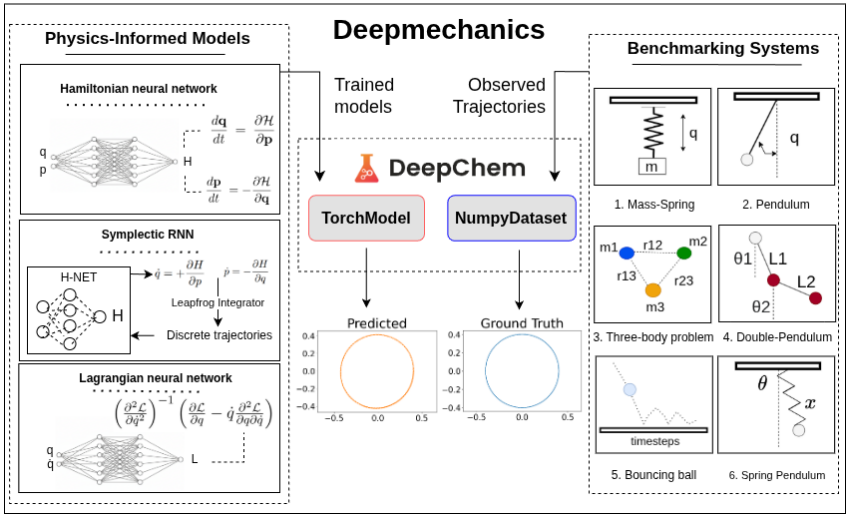
Preprint
Deepmechanics
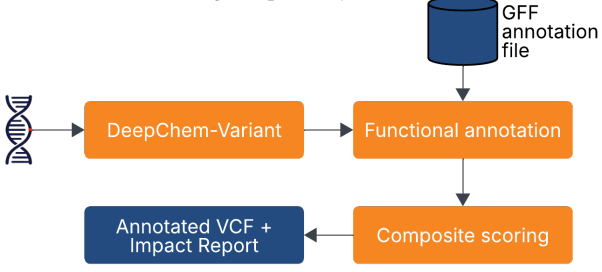
Preprint
AgriVariant: Variant Effect Prediction using DeepChem-Variant for Precision Breeding in Rice
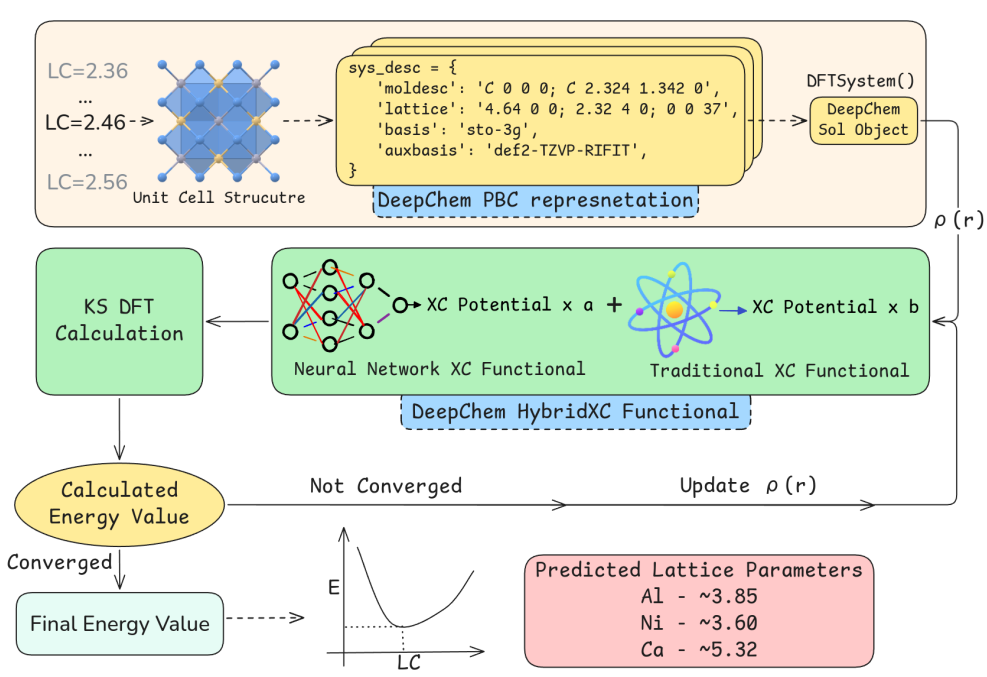
Preprint
A fully differentiable framework for training proxy Exchange Correlation Functionals for periodic systems
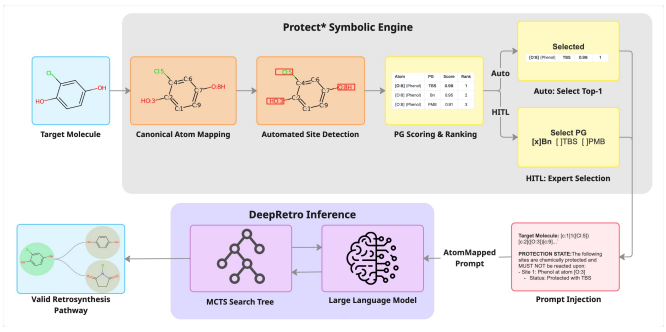
Preprint
Protect*: Steerable Retrosynthesis through Neuro-Symbolic State Encoding
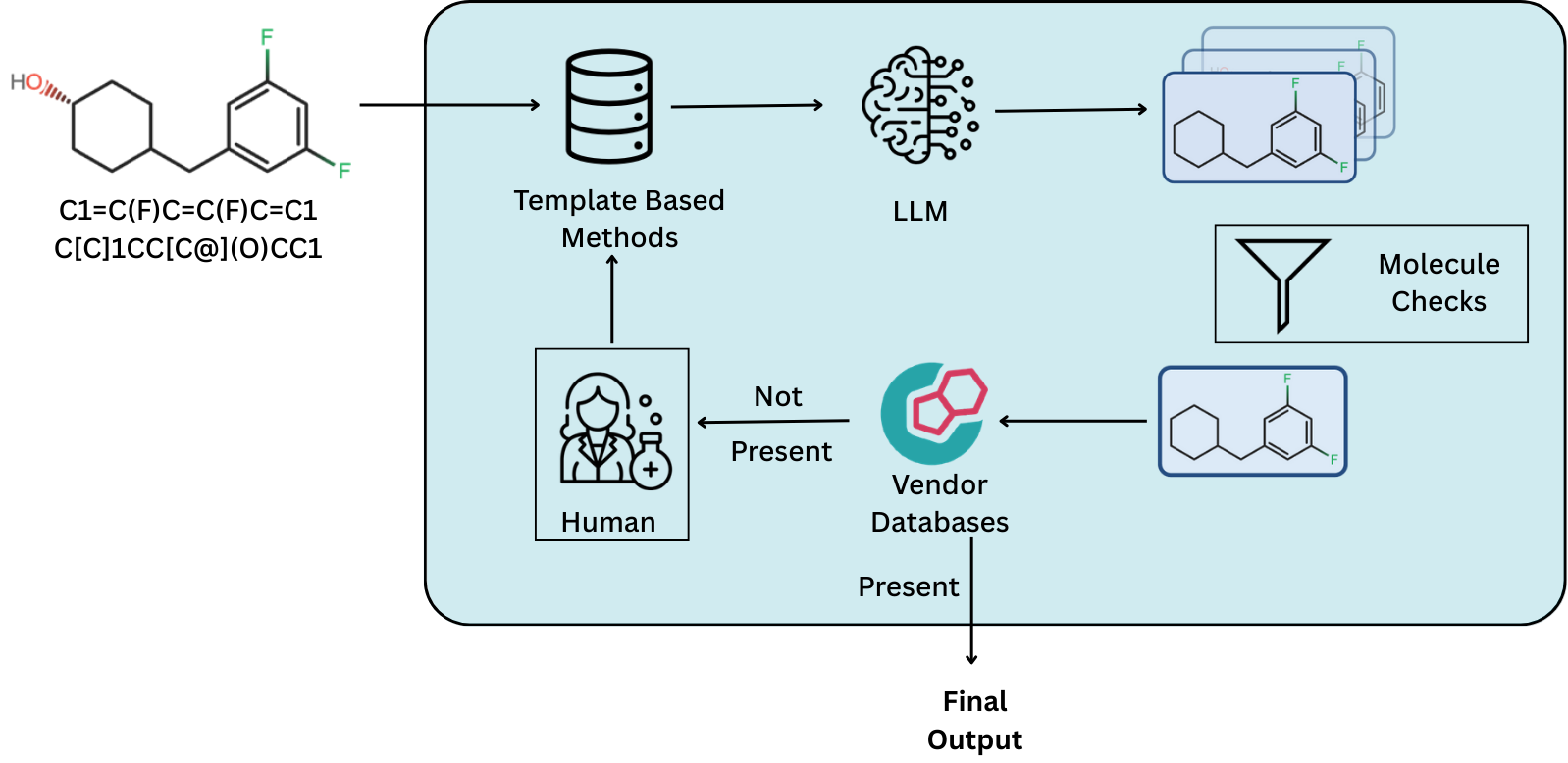
Journal Publication
DeepRetro discovers retrosynthetic pathways through iterative large language model reasoning
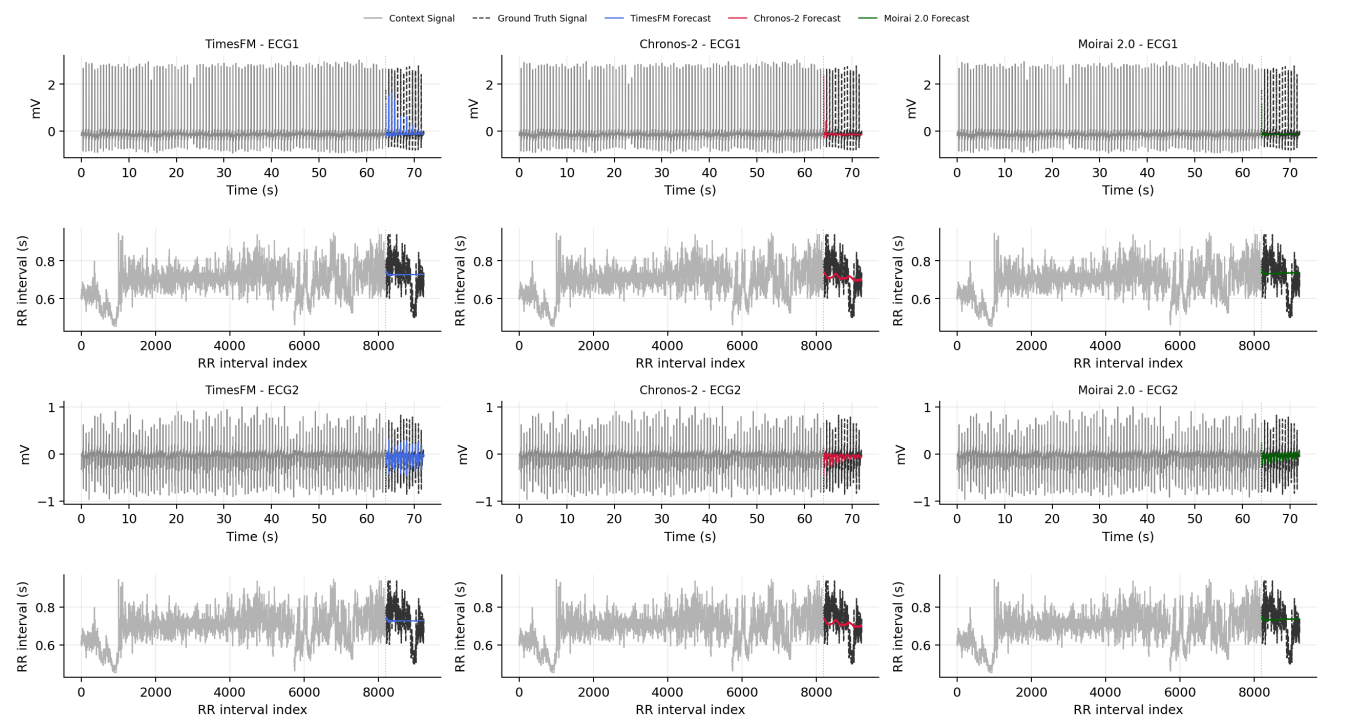
Preprint
Evaluating Long-Range Temporal Structure in Foundation Model-Based Forecasts of Heartbeat Dynamics
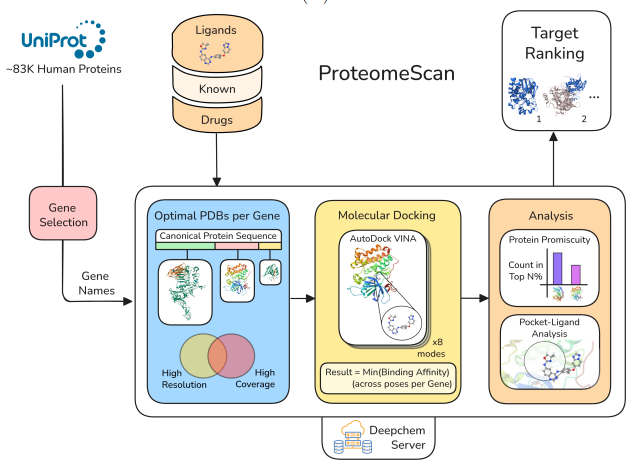
Preprint
ProteomeScan: A Toolkit For Target Validation By Proteome-Wide Docking And Analysis
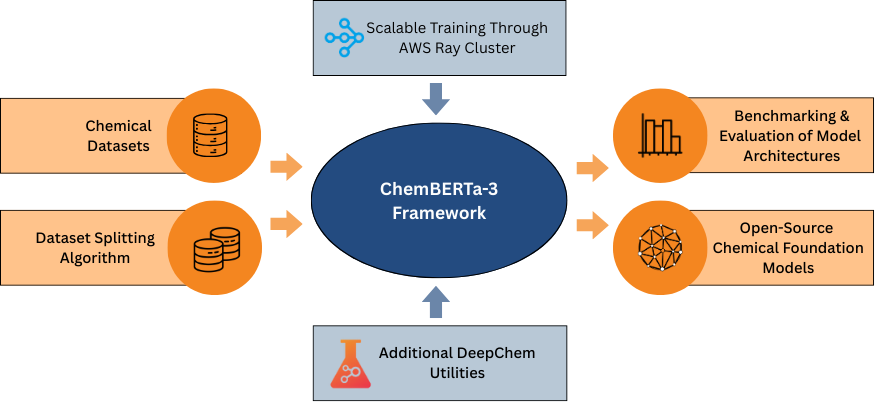
Journal Publication
ChemBERTa-3: an open source training framework for chemical foundation models
2025
Preprint
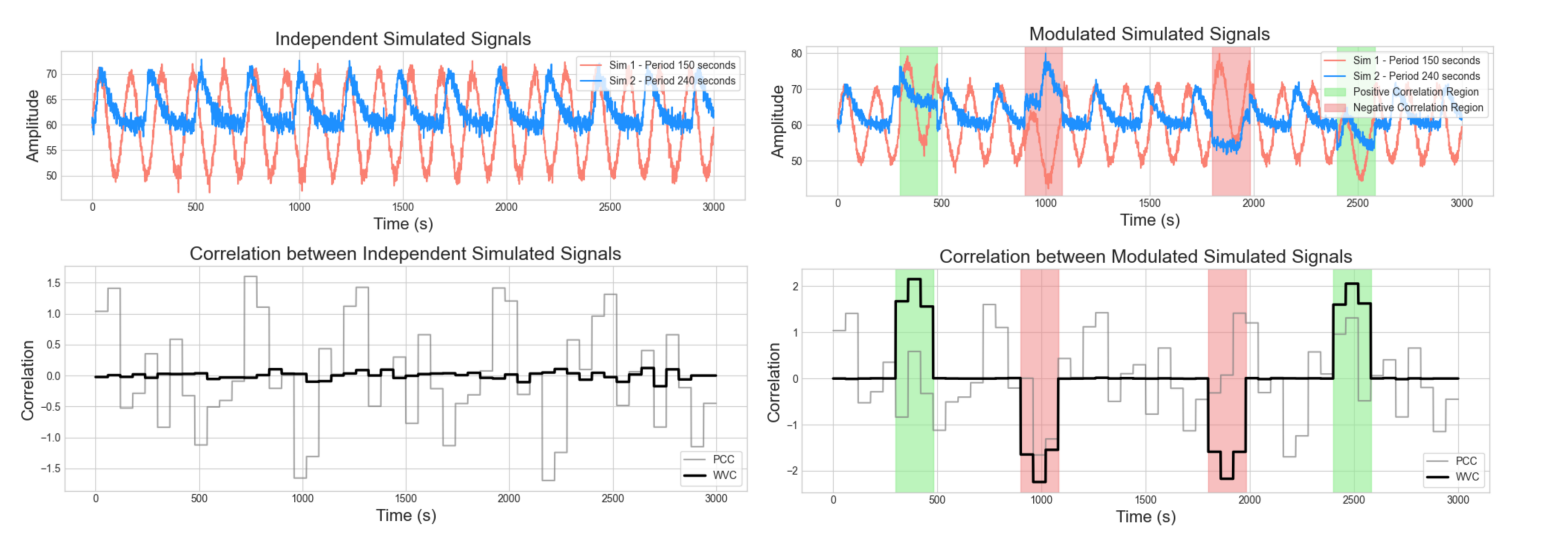
Inferring Dynamic Hidden Graph Structure in Heterogeneous Correlated Time Series
Preprint
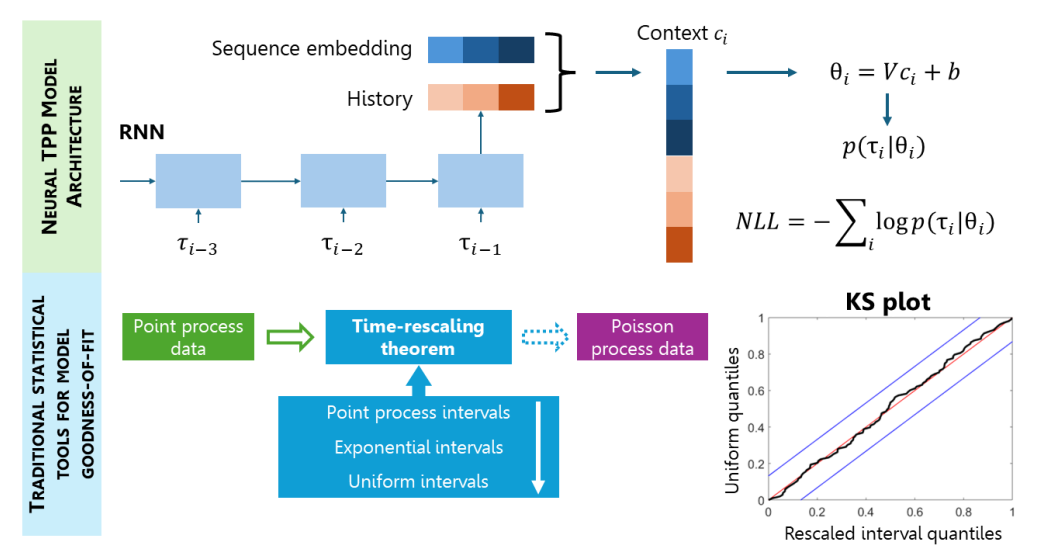
Density-based Neural Temporal Point Processes for Heartbeat Dynamics
Preprint
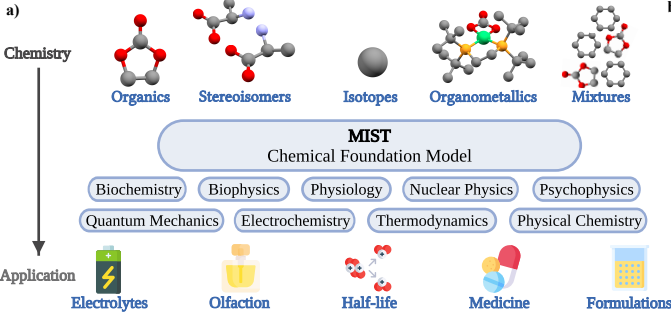
Foundation models for discovery and exploration in chemical space
Preprint
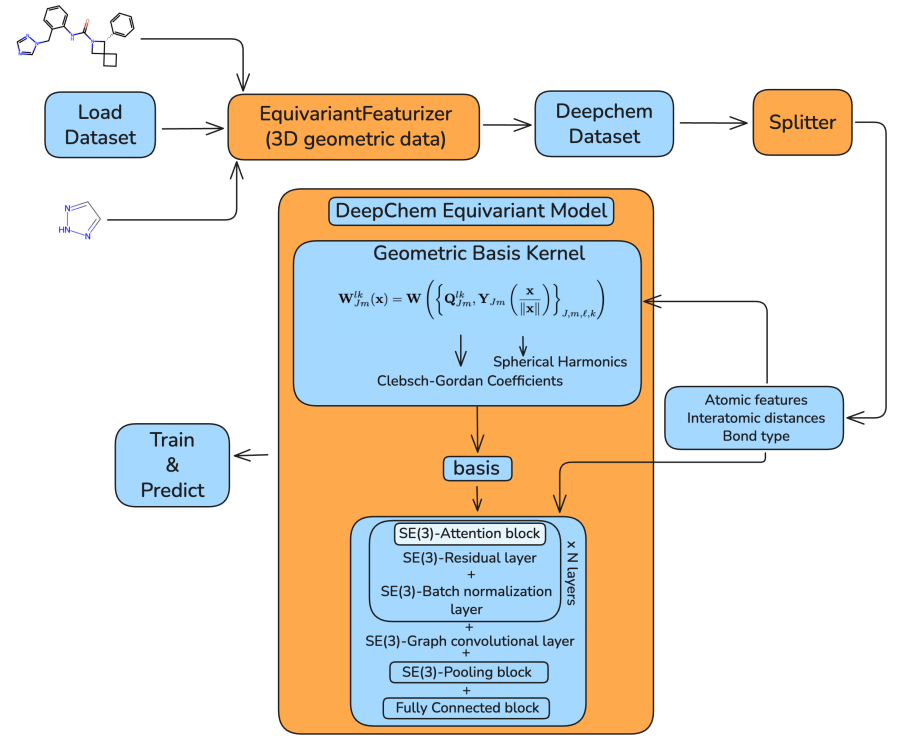
DeepChem Equivariant: SE (3)-Equivariant Support in an Open-Source Molecular Machine Learning Library
Preprint
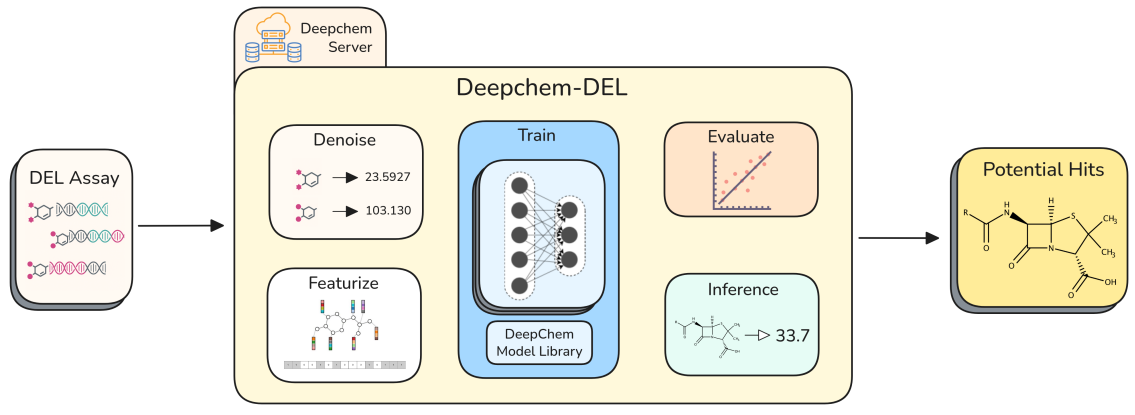
DeepChem-DEL: An Open Source Framework for Reproducible DEL Modeling and Benchmarking
Peer-reviewed Conference Publication
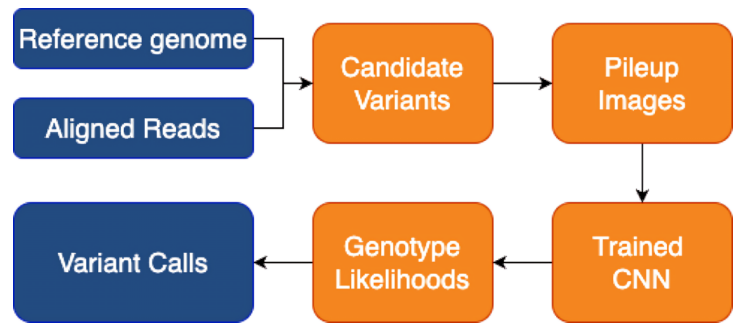
DeepChem-Variant: A Modular Open Source Framework for Genomic Variant Calling
Patent
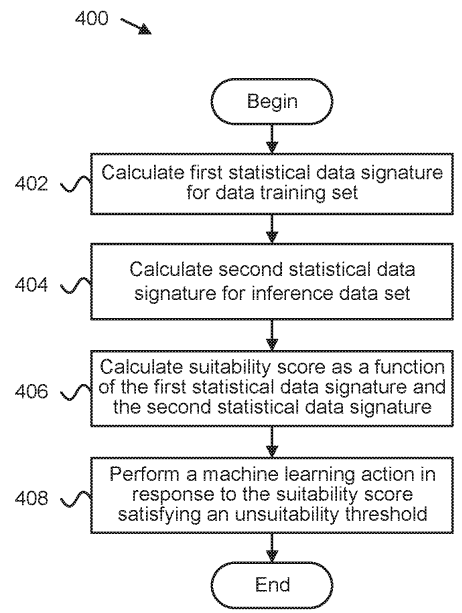
Detecting suitability of machine learning models for datasets.
Preprint
DeepRetro: Retrosynthetic Pathway Discovery using Iterative LLM Reasoning.
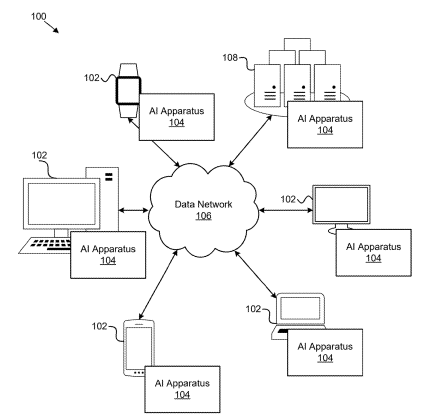
Patent
Ai-based drug side effect prediction.
Patent
Systems and methods for spatial graph convolutions with applications to drug discovery and molecular simulation.
2024
Preprint
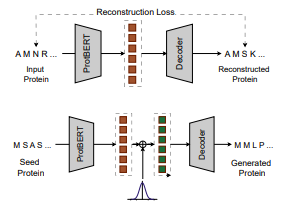
Open-Source Protein Language Models for Function Prediction and Protein Design.
Preprint
Open-source Polymer Generative Pipeline.
Preprint
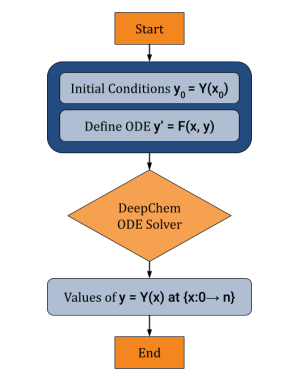
Open source Differentiable ODE Solving Infrastructure.
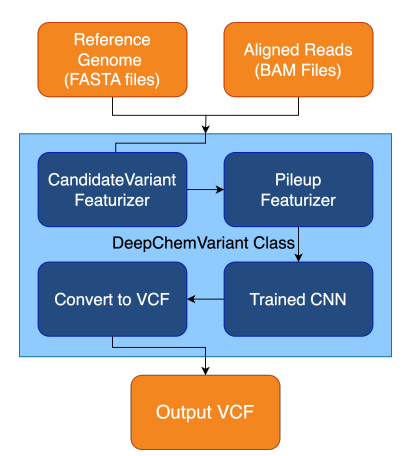
Preprint
A Modular Open Source Framework for Genomic Variant Calling.

Preprint
Open Source Infrastructure for Automatic Cell Segmentation.
Preprint
Self-supervised Pretraining for Partial Differential Equations
Peer-Reviewed Conference Publications
Open-Source Molecular Processing Pipeline for Generating Molecules
Peer-Reviewed Conference Publications
Machine Learning-Driven Predictions for Janus Kinase 3 Protein Drug Effectiveness
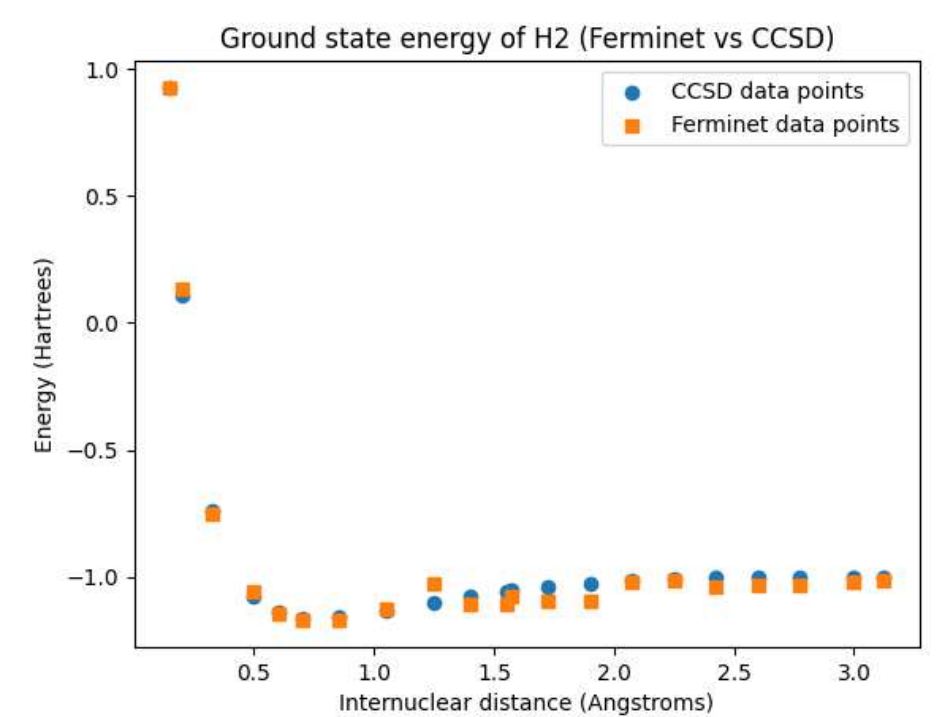
Peer-Reviewed Conference Publications
Open Source Fermionic Neural Networks with Ionic Charge Initialization
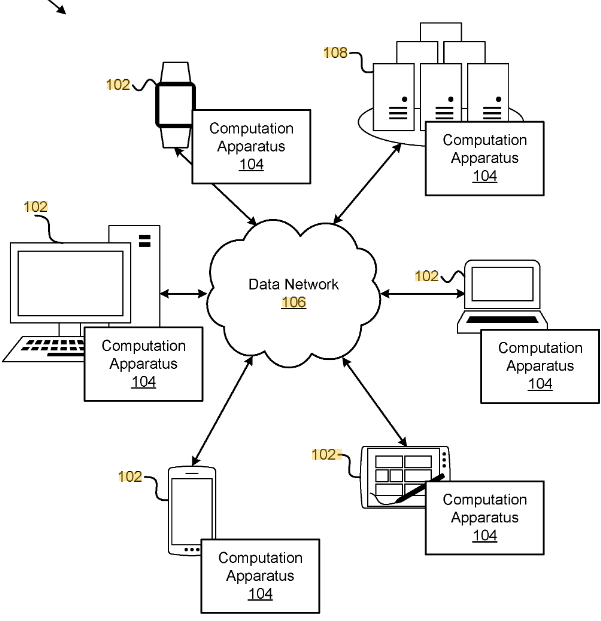
Patent
Techniques for a cloud scientific machine learning programming environment
2023
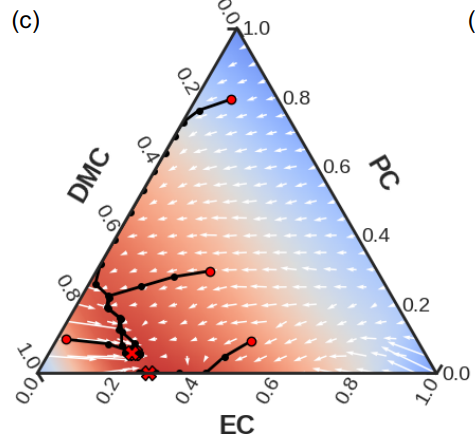
Journal Publication
Differentiable Modeling and Optimization of Battery Electrolyte Mixtures Using Geometric Deep Learning.
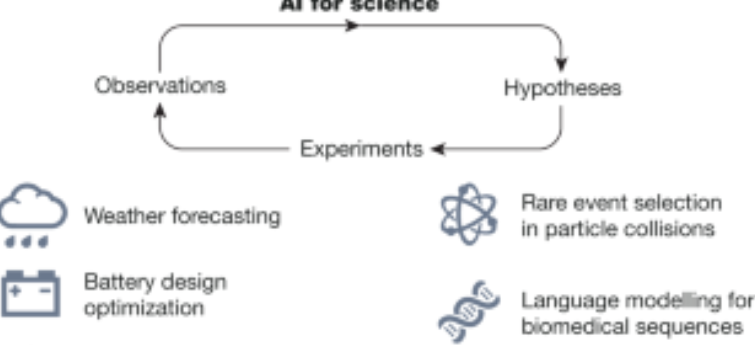
Journal Publication
Scientific discovery in the age of artificial intelligence.
Peer-Reviewed Conference Publications
Building AI Models of Patient-specific Drug Side Effect Predictions
Peer-Reviewed Conference Publications
Open Source Infrastructure for Differentiable Density Functional Theory
Peer-Reviewed Conference Publications
Score Based Models for Molecule Generation
Patent
Determining suitability of machine learning models for datasets
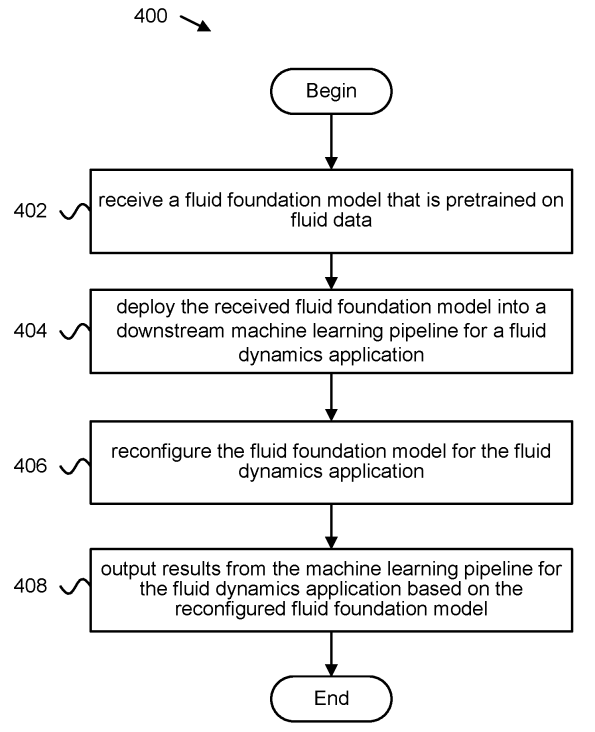
Patent
Foundation model based fluid simulations
2022
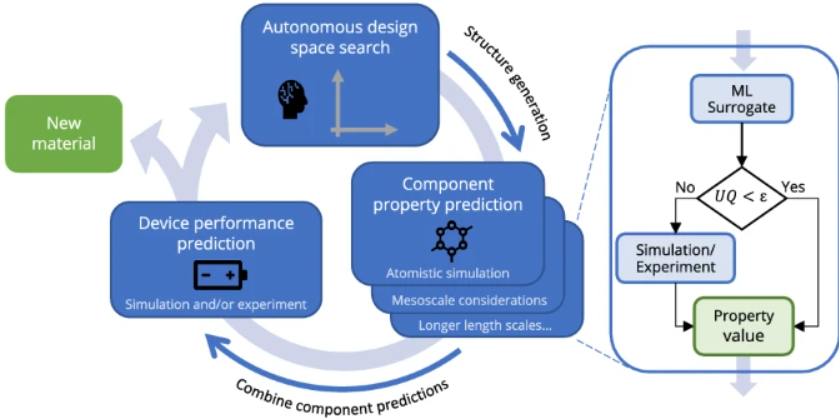
Journal Publication
AutoMat: Automated materials discovery for electrochemical systems.
Preprint
ChemBERTa-2: Towards Chemical Foundation Models
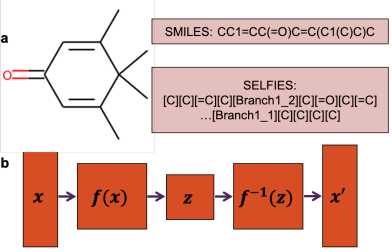
Peer-Reviewed Conference Publications
FastFlows: Flow-based Models for Molecular Graph Generation
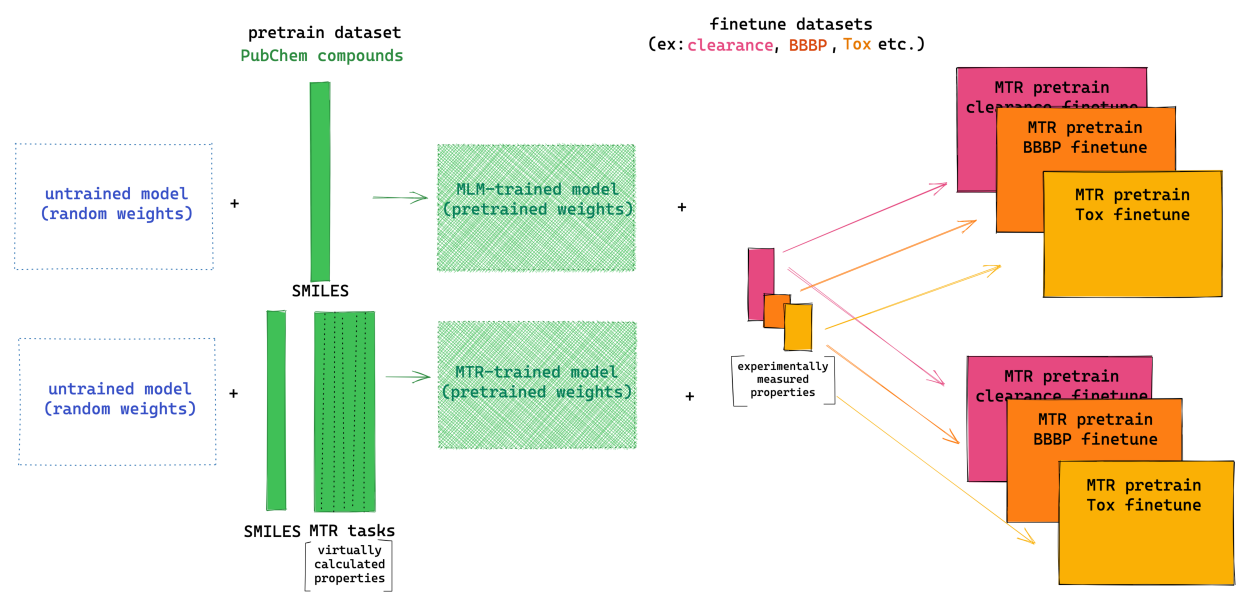
Peer-Reviewed Conference Publications
ChemBERTa-2: Towards Chemical Foundation Models
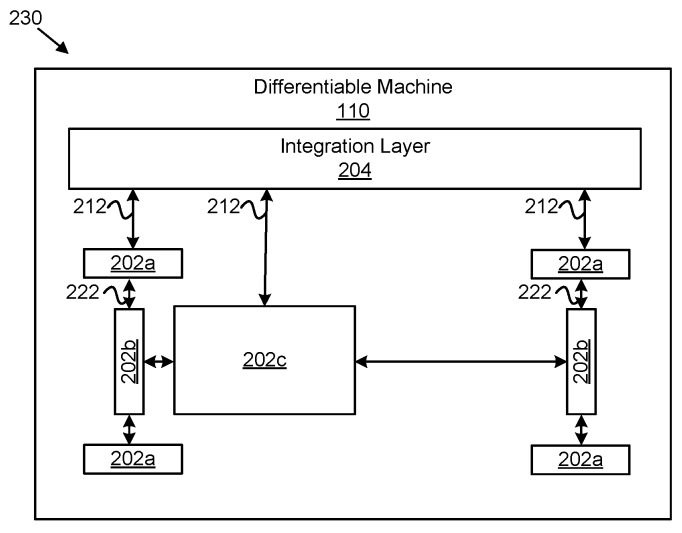
Patent
Differentiable machines for physical systems
2021
Preprint
Differentiable Physics: A Position Piece
Peer-Reviewed Conference Publications
Bringing Atomistic Deep Learning to Prime Time
2020
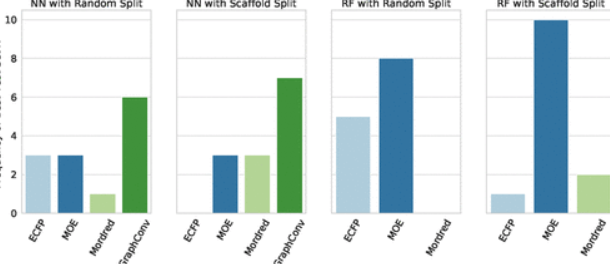
Journal Publication
AMPL: A Data-Driven Modeling Pipeline for Drug Discovery.

Preprint
SARS-CoV-2 and COVID-19: An Evolving Review of Diagnostics and Therapeutics.
Peer-Reviewed Conference Publications
ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction
Peer-Reviewed Conference Publications
Flow Based Models for Active Molecular Graph Generation
Patent
Interpretability-based machine learning adjustment during production
Patent
Determining validity of machine learning algorithms for datasets
2019
Journal Publication
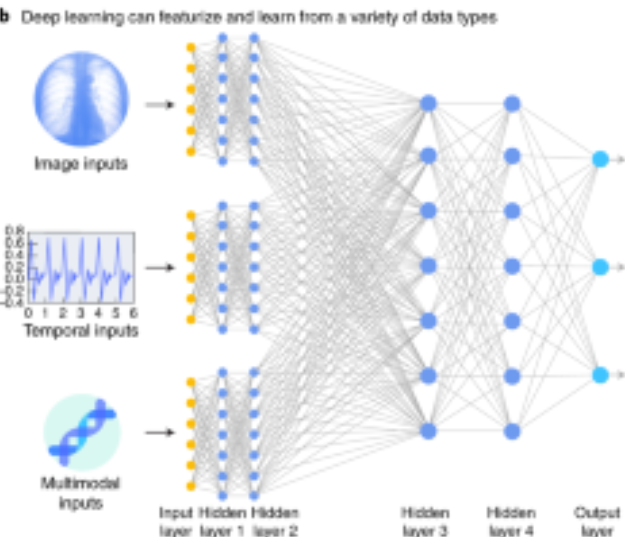
A guide to deep learning in healthcare.
Preprint
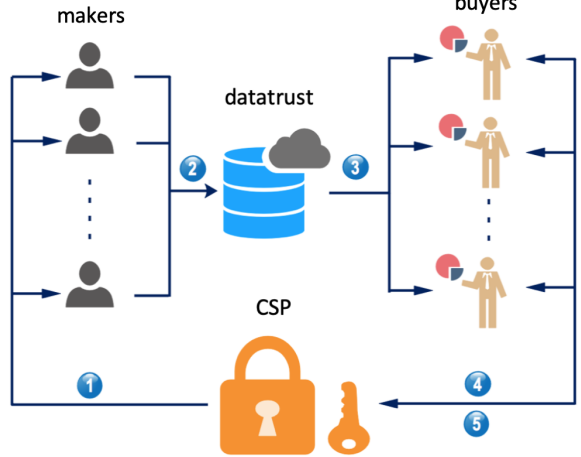
Secure Computation in Decentralized Data Markets
Patent
Detecting suitability of machine learning models for datasets
Patent
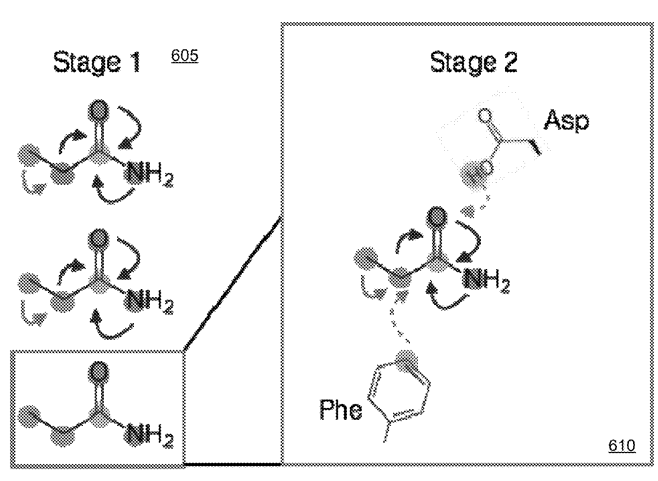
Systems and Methods for Spatial Graph Convolutions with Applications to Drug Discovery and Molecular Simulation
Patent
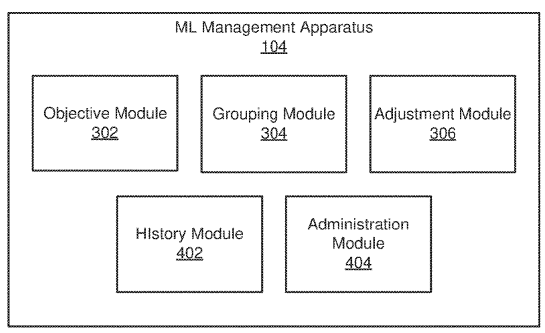
Machine learning abstraction
2018

Journal Publication
MoleculeNet: a benchmark for molecular machine learning
Journal Publication
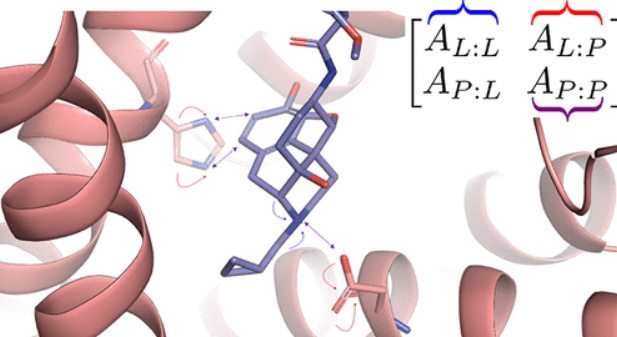
PotentialNet for molecular property prediction.
Journal Publication
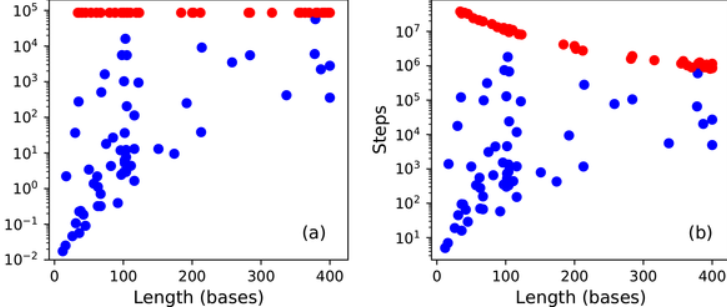
Solving the RNA design problem with reinforcement learning
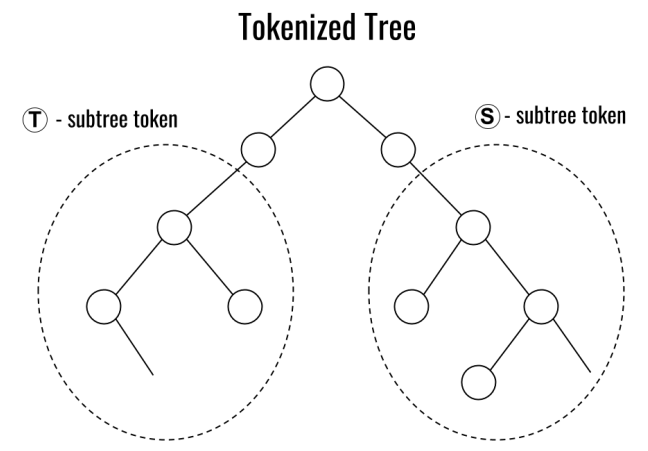
Preprint
Tokenized Data Markets
2017
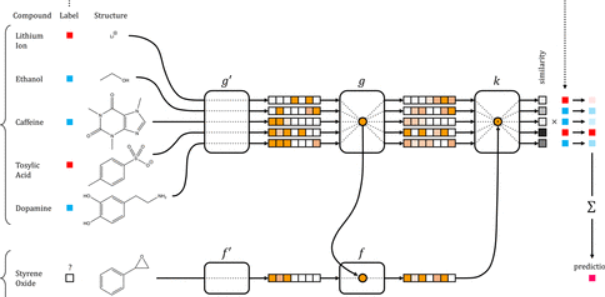
Journal Publication
Low Data Drug Discovery with One-Shot Learning
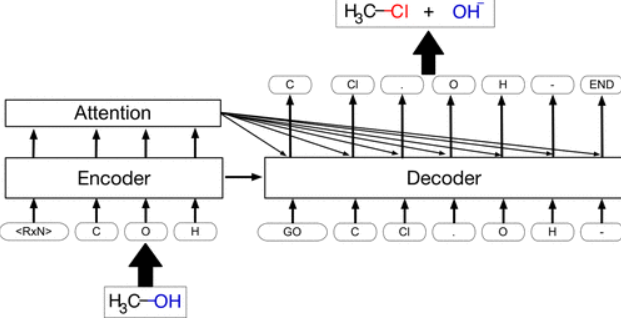
Journal Publication
Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models.
Journal Publication
Is Multitask Deep Learning Practical for Pharma?
2016
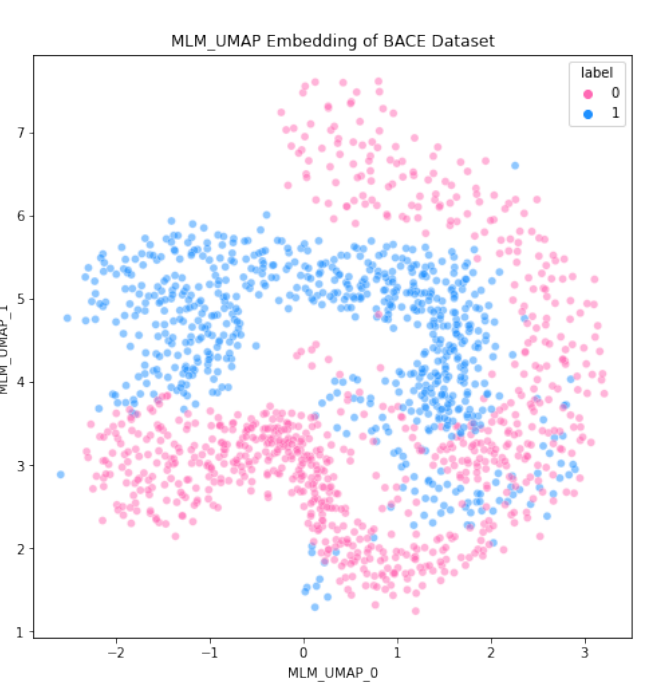
Journal Publication
Computational modeling of β-secretase 1 (BACE-1) inhibitors using ligand based approaches
Preprint
Learning Protein Dynamics with Metastable Switching Systems
Patent
Conditional iteration for a non-volatile device
2015
Preprint
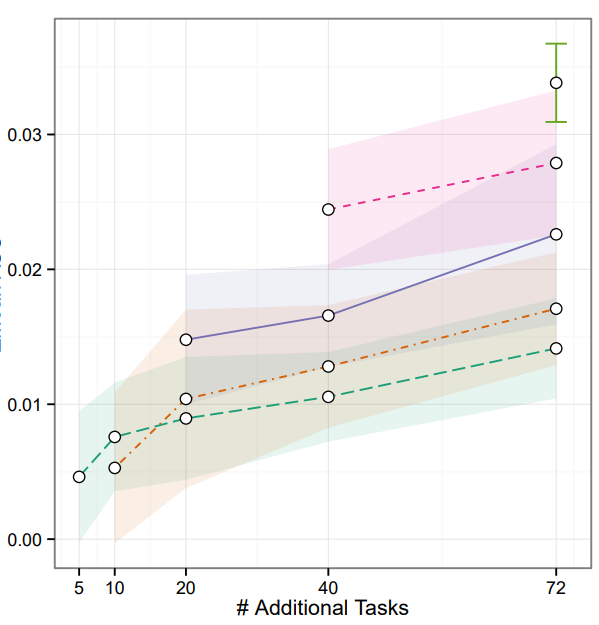
Massively Multitask Networks for Drug Discovery
Patent
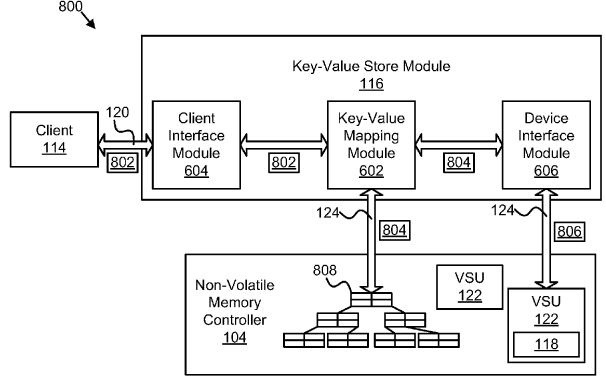
Non-volatile key-value store
2014
Peer-Reviewed Conference Publications
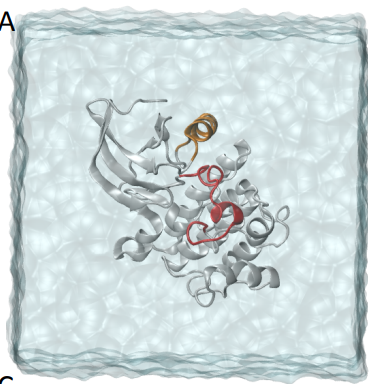
Understanding Protein Dynamics with L1-Regularized Reversible Hidden Markov Models
Peer-Reviewed Conference Publications
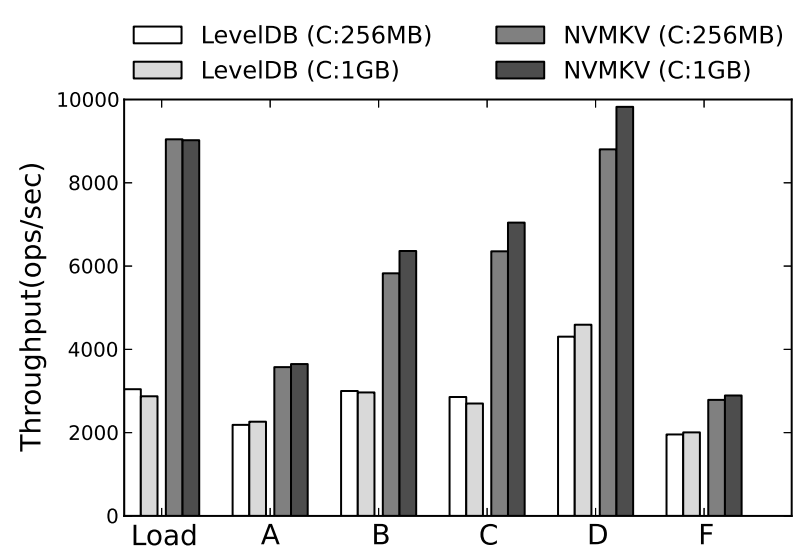
NVMKV: A Scalable and Lightweight Flash Aware Key-Value Store
2013
Peer-Reviewed Conference Publications
The extended parameter filter
Peer-Reviewed Conference Publications
Dynamic scaled sampling for deterministic constraints
Technical Blog Posts
- Mixture Descriptors toward the Development of Quantitative Structure–Property Relationship Models for the Flash Points of Organic Mixtures
- Lattice convolutional neural network modeling of adsorbate coverage effects
- Ultra-large library docking for discovering new chemotypes
- Deciphering interaction fingerprints from protein molecular surfaces
- Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties
- Unsupervised rational protein engineering with sequence only deep representation learning
- Induced Pluripotency as a Benchmark for Differentiable Cell Simulators
- Making Deep Learning Useful for Small Life Science Datasets
- Rethinking the Use of NLP Methods in the Life Sciences
Essays
- A PhD in Snapshots
- The Advent of Huang's Law
- Why Blockchain Could (One Day) Topple Google
- Learning to Learn
- What is Ethereum?
- Liquidity in Drug Discovery
- The Innovator's Open Source Dilemma
- Building The Open Source Drug Discovery Ecosystem
- What Can't Deep Learning Do?
- Machine Learning With Small Data
- Machine Learning With Small Data
- Software Patents Are Obsolete in the Age of AI
- தமிழில் விஞ்ஞானத்தை வளர்ப்போம்! (Science for all languages!)
- A Short Overview of Drug Discovery
- Why Should Drug Discovery Be Open Source?
- Software Is Eating Science
- Can AI Believe in God? A Parable about Diversity
- Learning Models of Disease
- The Ferocious Complexity of the Cell
- Why Antibiotics Are Hard
- Can Drugs Be Developed Like Open Source Projects